
|
| Home | : | Research | : | Publications | : | People | : | Lab Photos | : | Alumni | : | Contact | : | EBL | : | Software | : | Lab Wiki |
|
Publications
|
|
| We are making the the manuscripts listed on this page available to the academic community for use in teaching and research. The copyrights associated with each paper remain with the appropriate parties. | |
|
2019 |
|

|
Kaur H, Jamalidinan F, Condon SGF, Senes A, Hoskins AA
"Analysis of spliceosome dynamics by maximum likelihood fitting of dwell time distributions"
Methods 2019 153, 13-21
Abstract: Colocalization single-molecule methods can provide a wealth of information concerning the ordering and dynamics of biomolecule assembly. These have been used extensively to study the pathways of spliceosome assembly in vitro. Key to these experiments is the measurement of binding times-either the dwell times of a multi-molecular interaction or times in between binding events. By analyzing hundreds of these times, many new insights into the kinetic pathways governing spliceosome assembly have been obtained. Collections of binding times are often plotted as histograms and can be fit to kinetic models using a variety of methods. Here, we describe the use of maximum likelihood methods to fit dwell time distributions without binning. In addition, we discuss several aspects of analyzing these distributions with histograms and pitfalls that can be encountered if improperly binned histograms are used. We have automated several aspects of maximum likelihood fitting of dwell time distributions in the AGATHA software package. |
|
2018 |
|

|
Niemann MCE, Weber H, Hluska T, Leonte G, Anderson SM, Novak O, Senes A, Werner T
"The cytokinin oxidase/dehydrogenase CKX1 is a membrane-bound protein requiring homooligomerization in the endoplasmic reticulum for its cellular activity"
Plant Physiology 2018 176, 2024-39
Abstract: Degradation of the plant hormone cytokinin is controlled by cytokinin oxidase/dehydrogenase (CKX) enzymes. The molecular and cellular behavior of these proteins is still largely unknown. In this study, we show that CKX1 is a type-II single-pass membrane protein that predominantly localizes to the endoplasmic reticulum (ER). This indicates that this CKX isoform is a bona fide ER protein directly controlling the cytokinin which triggers the signaling from the ER. By using various approaches, we demonstrate that CKX1 forms homodimers and homooligomers in vivo. The N-terminal part of CKX1 was necessary and sufficient for the protein oligomerization as well as for targeting and retention in the ER. Moreover, we show that protein-protein interaction is largely facilitated by transmembrane helices and depends on a functional GxxxG-like interaction motif. Importantly, mutations rendering CKX1 monomeric interfere with its steady-state localization in the ER and cause a loss of the CKX1 biological activity by increasing its ER-associated degradation. Therefore, our study provides evidence that oligomerization is a crucial parameter regulating CKX1 biological activity and the cytokinin concentration in the ER. The work also lends a strong support for the cytokinin signaling from the ER and for the functional relevance of the cytokinin pool in this compartment. |

|
Condon SGF*, Mahbuba DA*, Armstrong CR, Diaz-Vazquez G, Craven SJ, LaPointe LM, Khadria AS, Chadda R, Crooks JA, Rangarajan N, Weibel DB, Hoskins AA, Robertson JL, Cui Q, Senes A
"The FtsLB sub-complex of the bacterial divisome is a tetramer with an uninterrupted FtsL helix linking the transmembrane and periplasmic regions"
J. Biol. Chem. 2018 293, 1623-41 (*authors contributed equally)
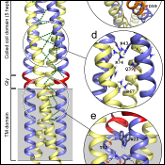
Abstract: In Escherichia coli, FtsLB plays a central role in the initiation of cell division, possibly transducing a signal that will eventually lead to the activation of peptidoglycan remodeling at the forming septum. The molecular mechanisms by which FtsLB operates in the divisome, however, are not understood. Here we present a structural analysis of the FtsLB complex — performed with biophysical, computational and in vivo methods — that establishes the organization of the transmembrane region and proximal coiled coil of the complex. FRET analysis in vitro is consistent with formation of a tetramer composed of two FtsL and two FtsB subunits. We predicted subunit contacts through co-evolutionary analysis and used them to compute a structural model of the complex. The transmembrane region of FtsLB is stabilized by hydrophobic packing and by a complex network of hydrogen bonds. The coiled coil domain likely terminates near the critical Constriction Control Domain, which might correspond to a structural transition. Presence of strongly polar amino acids within the core of the tetrameric coiled coil suggests that the coil may split into two independent FtsQ-binding domains. The helix of FtsB is interrupted between the transmembrane and coiled coil regions by a flexible Gly-rich linker. Conversely, the data suggest that FtsL forms an uninterrupted helix across the two regions, and that integrity of this helix is indispensable for the function of the complex. The FtsL helix is thus a candidate for acting as a potential mechanical connection to communicate conformational changes between periplasmic, membrane, and cytoplasmic regions. |
|
2017 |
|

|
Anderson SM*, Mueller BK*, Lange EJ and Senes A
"Combination of Cα-H hydrogen bonds and van der Waals packing modulates the stability of GxxxG-mediated dimers in membranes"
J Am Chem Soc. 2017 139, 15774-83 (*authors contributed equally)
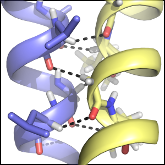
Abstract: The GxxxG motif is frequently found at the dimerization interface of a transmembrane structural motif called GASright, which is characterized by a short interhelical distance and a right-handed crossing angle between the helices. In GASright dimers, such as glycophorin A (GpA), BNIP3, and members of the ErbB family, the backbones of the helices are in contact, and they invariably display networks of 4 to 8 weak hydrogen bonds between Cα-H carbon donors and carbonyl acceptors on opposing helices (Cα-H···O=C hydrogen bonds). These networks of weak hydrogen bonds at the helix-helix interface are presumably stabilizing, but their energetic contribution to dimerization has yet to be determined experimentally. Here, we present a computational and experimental structure-based analysis of GASright dimers of different predicted stabilities, which show that a combination of van der Waals packing and Cα-H hydrogen bonding predicts the experimental trend of dimerization propensities. This finding provides experimental support for the hypothesis that the networks of Cα-H hydrogen bonds are major contributors to the free energy of association of GxxxG-mediated dimers. The structural comparison between groups of GASright dimers of different stabilities reveals distinct sequence as well as conformational preferences. Stability correlates with shorter interhelical distances, narrower crossing angles, better packing, and the formation of larger networks of Cα-H hydrogen bonds. The identification of these structural rules provides insight on how nature could modulate stability in GASright and finely tune dimerization to support biological function. |

|
Guo X, Niemi NM, Hutchins PD, Condon SGF, Jochem A, Ulbrich A, Higbee AJ, Russell JD, Senes A, Coon JJ, and Pagliarini DJ
"Ptc7p dephosphorylates select mitochondrial proteins to enhance metabolic function"
Cell Reports 2017 18, 307-13
Abstract: Proper maintenance of mitochondrial activity is essential for metabolic homeostasis. Widespread phosphorylation of mitochondrial proteins may be an important element of this process; yet little is known about which enzymes control mitochondrial phosphorylation, or which phosphosites have functional impact. We investigated these issues by disrupting Ptc7p — a conserved but largely uncharacterized mitochondrial matrix PP2C-type phosphatase. Loss of Ptc7p caused respiratory growth defects concomitant with elevated phosphorylation of select matrix proteins. Among these, Δptc7 yeast exhibited an increase in phosphorylation of Cit1p — the canonical citrate synthase of the tricarboxylic acid cycle — that diminished its activity. We found that phosphorylation of S462 can eliminate Cit1p enzymatic activity likely by disrupting its proper dimerization, and that Ptc7p-driven dephosphorylation rescued Cit1p activity. Collectively, our work connects Ptc7p to an essential TCA cycle function and to additional phosphorylation events that may affect mitochondrial activity inadvertently or in a regulatory manner. |
|
2016 |
|

|
Armstrong CR, Senes A
"Screening for transmembrane association in divisome proteins using TOXGREEN, a high-throughput variant of the TOXCAT assay"
Biochim. Biophys. Acta 2016 1858, 2573-83
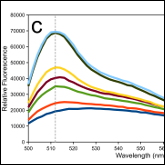
Abstract: TOXCAT is a widely used genetic assay to study interactions of transmembrane helices within the inner membrane of the bacterium Escherichia coli. TOXCAT is based on a fusion construct that links a transmembrane domain of interest with a cytoplasmic DNA-binding domain from the Vibrio cholerae ToxR protein. Interaction driven by the transmembrane domain results in dimerization of the ToxR domain, which, in turn, activates the expression of the reporter gene chloramphenicol acetyl transferase (CAT). Quantification of CAT is used as a measure of the ability of the transmembrane domain to self-associate. Because the quantification of CAT is relatively laborious, we developed a high-throughput variant of the assay, TOXGREEN, based on the expression of super-folded GFP and detection of fluorescence directly in unprocessed cell cultures. Careful side-by-side comparison of TOXCAT and TOXGREEN demonstrates that the methods have comparable response, dynamic range, sensitivity and intrinsic variability both in LB and minimal media. The greatly enhanced workflow makes TOXGREEN much more scalable and ideal for screening, since hundreds of constructs can be rapidly assessed in 96 well plates. Even for small scale investigations, TOXGREEN significantly reduces time, labor and cost associated with the procedure. We demonstrate applicability with a large screening for self-association among the transmembrane domains of bitopic proteins of the divisome (FtsL, FtsB, FtsQ, FtsI, FtsN, ZipA and EzrA) belonging to 11 bacterial species. The analysis confirms a previously reported tendency for FtsB to self-associate, and suggests that the transmembrane domains of ZipA, EzrA and FtsN may also possibly oligomerize. |

|
Barth P, Senes A
"Toward high-resolution computational design of the structure and function of helical membrane proteins"
Nat Struct Mol Biol 2016 23, 475-80
Abstract: The computational design of α-helical membrane proteins is still in its infancy but has already made great progress. De novo design allows stable, specific and active minimal oligomeric systems to be obtained. Computational reengineering can improve the stability and function of naturally occurring membrane proteins. Currently, the major hurdle for the field is the experimental characterization of the designs. The emergence of new structural methods for membrane proteins will accelerate progress. |

|
Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SGF, Lapolla SM, Hays, FA, Ding J, He F, Zhang XC, Li J, Senes A, Andrews DW, Lin J
"BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes"
EMBO J 2016 35, 208-36
Abstract: Pro-apoptotic Bax induces mitochondrial outer membrane permeabilization (MOMP) by forming oligomers through a largely undefined process. Using site-specific disulfide crosslinking, compartment-specific chemical labeling, and mutational analysis, we found that activated integral membrane Bax proteins form a BH3-in-groove dimer interface on the MOM surface similar to that observed in crystals. However, after the α5 helix was released into the MOM, the remaining interface with α2, α3, and α4 helices was rearranged. Another dimer interface was formed inside the MOM by two intersected or parallel α9 helices. Combinations of these interfaces generated oligomers in the MOM. Oligomerization was initiated by BH3-in-groove dimerization, without which neither the other dimerizations nor MOMP occurred. In contrast, α9 dimerization occurred downstream and was required for release of large but not small proteins from mitochondria. Moreover, the release of large proteins was facilitated by α9 insertion into the MOM and localization to the pore rim. Therefore, the BH3-in-groove dimerization on the MOM nucleates the assembly of an oligomeric Bax pore that is enlarged by α9 dimerization at the rim. |
|
2015 |
|

|
Khadria AS and Senes A
"Fluorophores, environments and quantification techniques in the analysis of transmembrane helix interaction using FRET"
Biopolymers 2015 104 (4), 247-264
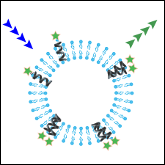
Abstract: FRET has been widely used as a spectroscopic tool in vitro to study the interactions between transmembrane helices in detergent and lipid environments. This technique has been instrumental to many studies that have greatly contributed to quantitative understanding of the physical principles that govern helix-helix interaction in the membrane. These studies have also improved our understanding of the biological role of oligomerization in membrane proteins. In this review, we focus on the combinations of fluorophores used, the membrane mimetic environments and measurement techniques that have been applied to study model systems as well as biological oligomeric complexes in vitro. We highlight the different formalisms used to calculate FRET efficiency, and the challenges associated with accurate quantification. The goal is to provide the reader with a comparative summary of the relevant literature for planning and designing FRET experiments aimed at measuring transmembrane helix-helix association. |

|
Ma G, Wei M, He L, Liu C, B Wu , Zhang S, Jing J, Liang X, Senes A, Tan P, Li S, Sun A, Bi Y, Zhong L, Si H, Shen Y, Li M, Lee MS, Zhou W, Wang J, Wang Y and Zhou Y
"Inside-out Ca2+ signaling prompted by STIM1 conformational switch"
Nat. Commun. 2015 6, 7826
Abstract: Store-operated Ca2+ entry mediated by STIM1 and ORAI1 constitutes one of the major Ca2+ entry routes in mammalian cells. The molecular choreography of STIM1-ORAI1 coupling is initiated by endoplasmic reticulum (ER) Ca2+ store depletion with subsequent oligomerization of the STIM1 ER-luminal domain, followed by its redistribution toward the plasma membrane to gate ORAI1 channels. The mechanistic underpinnings of this inside-out Ca2+ signaling were largely undefined. By taking advantage of a unique gain-of-function mutation within the STIM1 transmembrane domain (STIM1-TM), here we show that local rearrangement, rather than alteration in the oligomeric state of STIM1-TM, prompts conformational changes in the cytosolic juxtamembrane coiled coil region. Importantly, we further identify critical residues within the cytoplasmic domain of STIM1 (STIM1-CT) that entail autoinhibition. Our findings provide compelling evidence to support a conformational switch model in which STIM1-TM reorganization switches STIM1-CT into an extended conformation, thereby unleashing the ORAI-activating domain to gate ORAI1 channels. |
|
2014 |
|

|
Khadria AS, Mueller BK, Stefely JA, Tan CH, Pagliarini DJ and Senes A
"A Gly-zipper motif mediates homo-dimerization of the transmembrane domain of the mitochondrial kinase ADCK3"
J Am Chem Soc 2014 136, 14068-77
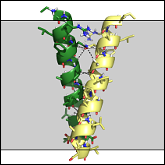
Abstract: Interactions between α-helices within the hydrophobic environment of lipid bilayers are integral to the folding and function of transmembrane proteins; however, the major forces that mediate these interactions remain debated, and our ability to predict these interactions is still largely untested. We recently demonstrated that the frequent transmembrane association motif GASright - the GxxxG-containing fold of the glycophorin A dimer - is optimal for the formation of extended networks of Cα-H hydrogen bonds, supporting the hypothesis that these bonds are major contributors to association. We also found that optimization of Cα-H hydrogen bonding and inter-helical packing is sufficient to computationally predict the structure of known GASright dimers at near atomic level. Here, we demonstrate that this computational method can be used to characterize the structure of a protein not previously known to dimerize, by predicting and validating the transmembrane dimer of ADCK3, a mitochondrial kinase. ADCK3 is involved in the biosynthesis of the redox active lipid, ubiquinone, and human ADCK3 mutations cause a cerebellar ataxia associated with ubiquinone deficiency, but the biochemical functions of ADCK3 remain largely undefined. Our experimental analyses show that the transmembrane helix of ADCK3 dimerizes, with an interface based on an extended Gly-zipper motif, as predicted by our models. The data provide strong evidence for the hypothesis that optimization of Cα-H hydrogen bonding is an important factor in the association of transmembrane helices. This work also provides a structural foundation for investigating the role of transmembrane association in regulating the biological activity of ADCK3. |

|
Subramaniam S and Senes A
"Backbone dependency further improves side chain prediction efficiency in the Energy-Based Conformer Library (bEBL)"
Proteins 2014 81 (11), 3177-3187
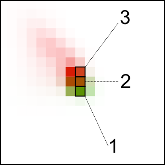
Abstract: Side chain optimization is an integral component of many protein modeling applications. In these applications, the conformational freedom of the side chains is often explored using libraries of discrete, frequently occurring conformations. Because side chain optimization can pose a computationally intensive combinatorial problem, the nature of these conformer libraries is important for ensuring efficiency and accuracy in side chain prediction. We have previously developed an innovative method to create a conformer library with enhanced performance. The Energy-Based Library (EBL) was obtained by analyzing the energetic interactions between conformers and a large number of natural protein environments from crystal structures. This process guided the selection of conformers with the highest propensity to fit into spaces that should accommodate a side chain. Because the method requires a large crystallographic data-set, the EBL was created in a backbone-independent fashion. However, it is well established that side chain conformation is strongly dependent on the local backbone geometry, and that backbone-dependent libraries are more efficient in side chain optimization. Here we present a backbone-dependent version of the EBL, the bEBL, whose conformers are independently sorted for each populated region of Ramachandran space. The resulting library closely mirrors the local backbone-dependent distribution of side chain conformation. Compared to the EBL, we demonstrate that the bEBL uses fewer conformers to produce similar side chain prediction outcomes, thus further improving performance with respect to the already efficient backbone-independent version of the library. |

|
Gromek KA, Suchy FP, Meddaugh HR, Wrobel RL, LaPointe LM, Chu UB, Primm JG, Ruoho AE, Senes A and Fox BG
"The Oligomeric States of the Purified Sigma 1 Receptor are Stabilized by Ligands"
J. Biol. Chem. 2014 289(29), 20333-44
Abstract: Sigma 1 receptor (S1R) is a mammalian member of the ERG2 and sigma1 receptor like protein family (pfam04622). It has been implicated in drug addiction and many human neurological disorders including Alzheimers and Parkinsons diseases and amyotrophic lateral sclerosis. A broad range of synthetic small molecules including cocaine, (+)-pentazocine, haloperidol and small endogenous molecules such as N,N-dimethyltryptamine, sphingosine and steroids have been identified as regulators of S1R. However, the mechanism of activation of S1R remains obscure. Here we provide evidence in vitro that S1R has ligand binding activity only in an oligomeric state. The oligomeric state is prone to decay into an apparent monomeric form when exposed to elevated temperature, with loss of ligand binding activity. This decay is suppressed in the presence of the known S1R ligands such as haloperidol, BD-1047 and sphingosine. S1R has a GxxxG motif in its second transmembrane region, and these motifs are often involved in oligomerization of membrane proteins. Disrupting mutations within the GxxxG motif shifted the fraction of the higher oligomeric states towards smaller states and resulted in a significant decrease in specific [3H]-(+)-pentazocine binding. Results presented here support the proposal that S1R function may be regulated by its oligomeric state. Possible mechanisms of molecular regulation of interacting protein partners by S1R in the presence of small molecule ligands are discussed. |

|
Mueller BK*, Subramaniam S* and Senes A
"A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα-H hydrogen bonds"
Proc Natl Acad Sci USA 2014 111(10) E888-95 (*authors contributed equally)
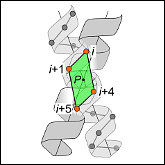
Abstract: Carbon hydrogen bonds between Cα-H donors and carbonyl acceptors are frequently observed between transmembrane helices (Cα-H···O=C). Networks of these interactions occur often at helix-helix interfaces mediated by GxxxG and similar patterns. Cα-H hydrogen bonds have been hypothesized to be important in membrane protein folding and association, but evidence that they are major determinants of helix association is still lacking. Here we present a comprehensive geometric analysis of homodimeric helices that demonstrates the existence of a single region in conformational space with high propensity for Cα-H···O=C hydrogen bond formation. This region corresponds to the most frequent motif for parallel dimers, GASright, whose best-known example is glycophorin A. The finding suggests a causal link between the high frequency of occurrence of GASright and its propensity for carbon hydrogen bond formation. Investigation of the sequence dependency of the motif determined that Gly residues are required at specific positions where only Gly can act as a donor with its "side chain" Hα. Gly also reduces the steric barrier for non-Gly amino acids at other positions to act as Cα donors, promoting the formation of cooperative hydrogen bonding networks. These findings offer a structural rationale for the occurrence of GxxxG patterns at the GASright interface. The analysis identified the conformational space and the sequence requirement of Cα-H···O=C mediated motifs; we took advantage of these results to develop a structural prediction method. The resulting program, CATM, predicts ab initio the known high-resolution structures of homodimeric GASright motifs at near-atomic level. |
|
2013 |
|

|
Khadria AS and Senes A
"The transmembrane domains of the bacterial cell division proteins FtsB and FtsL form a stable high-order oligomer"
Biochemistry 2013 52, 7532-41
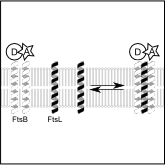
Abstract: FtsB and FtsL are two essential integral membrane proteins of the bacterial division complex or "divisome", both characterized by a single transmembrane helix and a juxtamembrane coiled coil domain. The two domains are important for the association of FtsB and FtsL, a key event for their recruitment to the divisome, which in turn allows the recruitment of the late divisomal components to the Z-ring and subsequent completion of the division process. Here we present a biophysical analysis performed in vitro that shows that the transmembrane domains of FtsB and FtsL associate strongly in isolation. Using Förster resonance energy transfer, we have measured the oligomerization of fluorophore-labeled transmembrane domains of FtsB and FtsL in both detergent and lipid. The data indicate that the transmembrane helices are likely a major contributor to the stability of the FtsB−FtsL complex. Our analyses show that FtsB and FtsL form a 1:1 higher-order oligomeric complex, possibly a tetramer. This finding suggests that the FtsB−FtsL complex is capable of multivalent binding to FtsQ and other divisome components, a hypothesis that is consistent with the possibility that the FtsB−FtsL complex has a structural role in the stabilization of the Z-ring. |

|
Khadria A and Senes A
"Measurement of transmembrane peptide interaction in liposomes using Forster Resonance Energy Transfer (FRET)"
Methods Mol. Biol. 2013 1063, 19-36
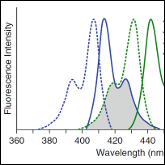
Abstract: Present day understanding of the thermodynamic properties of integral membrane proteins (IMPs) lags behind that of water-soluble proteins due to difficulties in mimicking the physiological environment of the IMPs in order to obtain a reversible folded system. Despite such challenges faced in studying these systems, significant progress has been made in the study of the oligomerization of single span transmembrane helices. One of the primary methods available to characterize these systems is based on Förster Resonance Energy Transfer (FRET). FRET is a widely used spectroscopic tool that provides proximity data that can be fitted to obtain the energetics of a system. Here we discuss various technical aspects related to the application of FRET to study transmembrane peptide oligomerization in liposomes. The analysis is based on FRET efficiency relative to the concentration of the peptides in the bilayer (peptide:lipid ratio). Some important parameters that will be discussed include labeling efficiency, sample homogeneity and equilibration. Furthermore, data analysis has to be performed keeping in mind random colocalization of donors and acceptors in liposome vesicles. |

|
LaPointe LM, Taylor KC, Subramaniam S, Khadria A, Rayment I and Senes A
"Structural organization of FtsB, a transmembrane protein of the bacterial divisome"
Biochemistry 2013 52, 2574-85
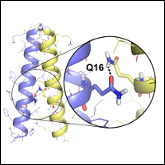
Abstract: We report the first structural analysis of an integral membrane protein of the bacterial divisome. FtsB is a single-pass membrane protein with a periplasmic coiled coil. Its heterologous association with its partner FtsL represents an essential event for the recruitment of the late components to the division site. Using a combination of mutagenesis, computational modeling and X-ray crystallography, we determined that FtsB self-associates and we investigated its structural organization. We found that the transmembrane domain of FtsB homo-oligomerizes through an evolutionarily conserved interaction interface where a polar residue (Gln 16) plays a critical role through the formation of an inter-helical hydrogen bond. The crystal structure of the periplasmic domain, solved as a fusion with Gp7, shows that 30 juxta-membrane amino acids of FtsB form a canonical coiled coil. The presence of conserved Gly residue in the linker region suggests that flexibility between the transmembrane and coiled coil domains is functionally important. We hypothesize that the transmembrane helices of FtsB form a stable dimeric core for its association with FtsL into a higher-order oligomer, and that FtsL is required to stabilize the periplasmic domain of FtsB, leading to the formation of a complex that is competent for binding to FtsQ, and to their consequent recruitment to the divisome. The study provides an experimentally validated structural model and identifies point mutations that disrupt association, thereby establishing important groundwork for the functional characterization of FtsB in vivo. |
|
2012 |
|

|
Subramaniam S and Senes A
"An Energy-Based conformer library for side chain optimization: improved prediction and adjustable sampling"
Proteins 2012 80, 2218-34
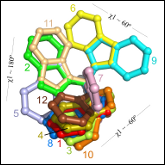
Abstract: Side chain optimization is a fundamental component of protein modeling applications such as docking, structural prediction, and design. In these applications side chain flexibility is often provided by rotamer or conformer libraries, which are collections of representative side chain conformations. Here we demonstrate that the sampling provided by the library can be substantially improved by adding an energetic criterion to its creation. The result of the new procedure is the Energy-Based library, a conformer library selected according to the propensity of its elements to fit energetically into natural protein environments. The new library performs outstandingly well in side chain optimization, producing structures with significantly lower energies and resulting in improved side chain conformation prediction. In addition, because the library was created as an ordered list, its size can be adjusted to any desired level. This feature provides unprecedented versatility in tuning sampling. It allows to precisely balance the number of conformers required by each amino acid type, equalizing their chances to fit into structural environments. It also allows to scale the amount of sampling to the specific requirement of any given side optimization problem. A rotameric version of the library was also produced with the same method to support applications that require a dihedral-only description of side chain conformation. The libraries are available at http://seneslab.org/EBL. |

|
Kulp DW, Subramaniam S, Donald JE, Hannigan BT, Mueller BK, Grigoryan G and Senes A
"Structural informatics, modeling and design with an open-source Molecular Software Library (MSL)"
J Comput. Chem. 2012 33(20), 1645-61
Abstract: We present the Molecular Software Library (MSL), a C++ library for molecular modeling. MSL is a set of tools that supports a large variety of algorithms for the design, modeling, and analysis of macromolecules. Among the main features supported by the library are methods for applying geometric transformations and alignments, the implementation of a rich set of energy functions, side chain optimization, backbone manipulation, calculation of solvent accessible surface area, and other tools. MSL has a number of unique features, such as the ability of storing alternative atomic coordinates (for modeling) and multiple amino acid identities at the same backbone position (for design). It has a straightforward mechanism for extending its energy functions and can work with any type of molecules. Although the code base is large, MSL was created with ease of developing in mind. It allows the rapid implementation of simple tasks while fully supporting the creation of complex applications. Some of the potentialities of the software are demonstrated here with examples that show how to program complex and essential modeling tasks with few lines of code. MSL is an ongoing and evolving project, with new features and improvements being introduced regularly, but it is mature and suitable for production and has been used in numerous protein modeling and design projects. MSL is open-source software, freely downloadable at http://msl-libraries.org. We propose it as a common platform for the development of new molecular algorithms and to promote the distribution, sharing, and reutilization of computational methods. |
|
2011 |
|

|
Hsin J, LaPointe LM, Kazy A, Chipot CJ, Senes A, Schulten K
"Oligomerization State of Photosynthetic Core Complexes is Correlated with the Dimerization Affinity of a Transmembrane Helix"
J Am Chem Soc. 2011 133(35), 14071-81
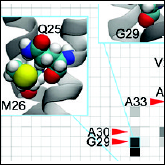
Abstract: In the Rhodobacter (Rba.) species of photosynthetic purple bacteria, a single transmembrane α-helix, PufX, is found within the core complex, an essential photosynthetic macromolecular assembly that performs the absorption and the initial processing of light energy. Despite its structural simplicity, many unresolved questions surround PufX, the most important of which is its location within the photosynthetic core complex. One proposed placement of PufX is at the center of a core complex dimer, where two PufX helices associate in the membrane and form a homodimer. Inability for PufX of certain Rba. species to form a homodimer is thought to lead to monomeric core complexes. In the present study, we employ a combination of computational and experimental techniques to test the hypothesized homodimerization of PufX. We carry out a systematic investigation to measure the dimerization affinity of PufX from four Rba. species, Rba. blasticus , Rba. capsulatus , Rba. sphaeroides , and Rba. veldkampii , using a molecular dynamics-based free-energy method, as well as experimental TOXCAT assays. We found that the four PufX helices have substantially different dimerization affinities. Both computational and experimental techniques demonstrate that species with dimeric core complexes have PufX that can potentially form a homodimer, whereas the one species with monomeric core complexes has a PufX with little to no dimerization propensity. Our analysis of the helix-helix interface revealed a number of positions that may be important for PufX dimerization and the formation of a hydrogen-bond network between these GxxxG-containing helices. Our results suggest that the different oligomerization states of core complexes in various Rba. species can be attributed, among other factors, to the different propensity of its PufX helix to homodimerize. |

|
Senes A
"Computational Design of Membrane Proteins"
Curr Opin Struct Biol. 2011 21(4), 460-6
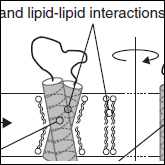
Abstract: This article reviews the recent successes of computational protein design techniques applied to integral membrane proteins. This emerging area is still handicapped by significant difficulties in the experimental characterization of the stability and structure of the designed proteins. Nevertheless, by focusing on oligomeric complexes of single-span transmembrane (TM) peptides with detectable activity, the computational design of membrane proteins has already produced very exciting results. The 'take-home message' is that optimization of van der Waals packing and hydrogen bonding (both 'canonical' and weak Cα-H⋯O bonds) can produce functional structures of remarkable stability and specificity in the membrane. |

|
Subramaniam S, Natarajan S, and Senes A
"A Machine Learning based Approach to Improve Protein Sidechain Optimization"
ACM Conference on Bioinformatics, Computational Biology and Biomedicine (ACM BCB) 2011, 478-480
Abstract: Side chain optimization is the process of packing the sidechains of a protein onto a fixed backbone structure, such that the energy of the resultant structure is minimized. The continuous space of sidechain conformations is typically handled by discretizing (sampling) into a finite set of representative conformations called a "conformer library". The key contribution of this work is to use machine learning methods to distribute (conformational) sampling among different positions in a protein. The idea is that different positions in a protein backbone have different sampling requirements, for example, solvent exposed positions require less sampling than positions in the core of a protein. We propose a 3-ary categorization of every position in a target protein based on its sampling requirements and evaluate it by comparing against an unbiased distribution of conformers. Our results demonstrate that this strategy helps to distribute the sampling more efficiently for sidechain optimization. |
|
Postdoctoral and graduate publications |
|
| Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, DeGrado, WF "De novo design and molecular assembly of a transmembrane diporphyrin-binding protein complex" J Am Chem Soc. 2010 132(44), 15516-8 | |
| Berger BW, Kulp DW, Span LM, DeGrado JL, Billings PC, Senes A, Bennett JS, DeGrado WF. "Consensus motif for integrin transmembrane helix association." Proc Natl Acad Sci USA/i> 2010 107(2), 703-8 | |
| McAllister KA, Zou H, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, Therien MJ, Blasie JK, DeGrado WF. "Using alpha-helical coiled-coils to design nanostructured metalloporphyrin arrays." J Am Chem Soc. 2008 130(36), 11921-7 | |
| Finikova OS, Troxler T, Senes A, DeGrado WF, Hochstrasser RM, Vinogradov SA. "Energy and electron transfer in enhanced two-photon-absorbing systems with triplet cores." J Phys Chem A. 2007 111(30), 6977-90 | |
| Senes A, Chadi DC, Law PB, Walters RF, Nanda V, DeGrado WF. "E(z), a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes, derivation and applications to determining the orientation of transmembrane and interfacial helices." J Mol Biol. 2007 366(2), 436-48 | |
| Fang C, Senes A, Cristian L, DeGrado WF, Hochstrasser RM. "Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer." Proc Natl Acad Sci USA 2006 103(45), 16740-5 | |
| Lehnert U, Xia Y, Royce TE, Goh CS, Liu Y, Senes A, Yu H, Zhang ZL, Engelman DM, Gerstein M. "Computational analysis of membrane proteins: genomic occurrence, structure prediction and helix interactions." Q Rev Biophys. 2004 37(2), 121-46 | |
| Senes A, Engel DE, DeGrado WF. "Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs." Curr Opin Struct Biol. 2004 14(4), 465-79 | |
| Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. "Membrane protein folding: beyond the two stage model." FEBS Lett. 2003 555(1), 122-5 | |
| Lin J, Qian J, Greenbaum D, Bertone P, Das R, Echols N, Senes A, Stenger B, Gerstein M. "GeneCensus, genome comparisons in terms of metabolic pathway activity and protein family sharing." Nucleic Acids Res. 2002 30(20), 4574-82 | |
| Senes A, Ubarretxena-Belandia I, Engelman DM. "The Calpha ---H...O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions." Proc Natl Acad Sci USA 2001 98(16), 9056-61 | |
| Senes A, Gerstein M, Engelman DM. "Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions." J Mol Biol. 2000 296(3), 921-36 | |
| Formato M, Senes A, Soccolini F, Coinu R, Cherchi GM. "A reversed phase HPLC method for the simultaneous determination of all monosaccharides contained in galactosaminoglycan isomers from human aorta proteoglycans." Carbohydr Res. 1994 255, 27-39 |
Associate Professor
Department of Biochemistry - UW-Madison
433 Backcock Dr., Room 419
Madison, WI 53706
office (+1) 608-890-2584
lab. (+1) 608-262-7355