
|
| Home | : | Research | : | Publications | : | People | : | Lab Photos | : | Alumni | : | Contact | : | EBL | : | Software | : | Lab Wiki |
|
Research: Structure, Prediction, and Design of Integral Membrane Proteins
|
||||||
|
We are interested in understanding structural properties of integral membrane proteins, with a particular focus on oligomeric complexes of proteins that have a single transmembrane domain.
Obtaining structural information for membrane proteins with the traditional methods (X-ray cryallography and NMR) is still difficult. Our general strategy is to combine biophysical and biochemical methods with computational modeling to interpret "low resolution" experimental information such as mutagenesis. Current projects
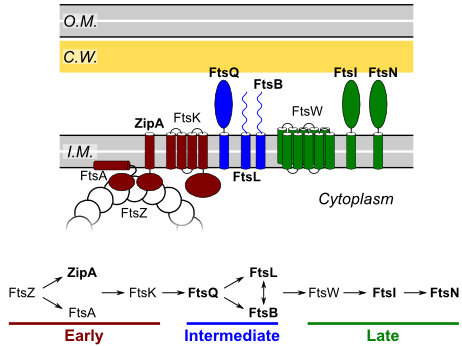
Structural studies of the transmembrane proteins of the bacterial divisome
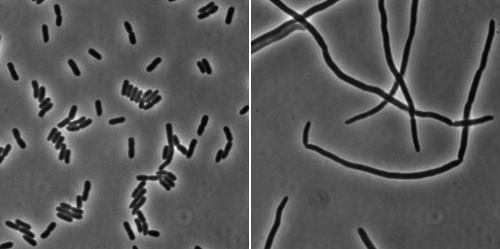
Cell division - one of the most fundamental and complex processes in the life of bacteria - requires mechanisms for the coordinated remodeling of the cell envelope, to control the synthesis of the septal cell wall, and to induce membrane fusion. These events are enabled by a membrane-localized complex. The components of the divisome have been identified and extensively analyzed in vivo, but little is known about the tridimentional structure of the complex. Our goal is to characterize the structural organization and interactions of the important transmembrane region, which was essentially unexplored.
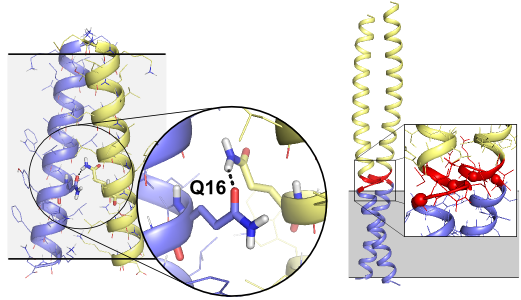
We are currently characterizing the structural organization of the complex of FtsB and FtsL, two single-pass proteins with a juxta-membrane coiled coil region. Using a combination of mutagenesis, molecular modeling and X-ray crystallography, we have recently determined that FtsB self-associates and obtained a structural model for both transmembrane and periplasmic region of the protein. (LaPointe LM, Taylor KC, Subramaniam S, Khadria A, Rayment I and Senes A Biochemistry 2013, in press)
We are currently investigating the biological role of these structural features using phenotypic and localization assays in vivo.
An Energy-based Conformer Library for Side Chain Prediction
A fundamental task in the prediction of protein structures is the correct placement of the flexible side chains. This process, called side-chain optimization, is most often performed using libraries of side chain conformation that cover the natural variability observed in protein structures. Because each individual position in a protein needs to be provided with multiple conformations, side-chain optimization poses a formidable combinatorial problem. Here we show that a library of conformations developed with a novel energy-based criterion allow us to use a smaller number of conformations, thus reducing computation time and at the same time improving the prediction results. MSL: an Open Source C++ toolbox for The Creation of Molecular Modeling Programs
We have created an open source C++ software library that supports the development of new programs and methods for the manipulation, analysis, modeling and design of proteins and other macromolecules. We propose it as a common platform for facilitating the distribution, sharing and reutilization of computational methods. MSL is available for free on the open source repository Source Forge. |
Associate Professor
Department of Biochemistry - UW-Madison
433 Backcock Dr., Room 419
Madison, WI 53706
office (+1) 608-890-2584
lab. (+1) 608-262-7355