Subramaniam S and Senes A
"An Energy-Based conformer library for side chain optimization: improved prediction and adjustable sampling"
Proteins 2012 in press DOI: 10.1002/prot.24111
Article : Pubmed : PDF
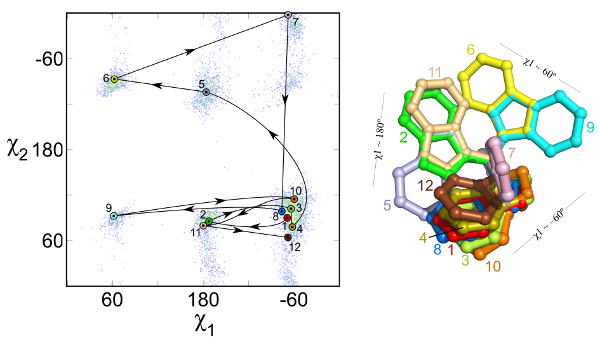
A major component of the prediction of protein structures is the correct placement of the flexible side chains. This process, called side-chain optimization, is most often performed using libraries of side chain conformation that cover the natural variability observed in protein structures. Because each individual position in a protein need to be provided with multiple conformations, side-chain optimization poses a formidable combinatorial problem. We have developed a new library with a novel energy-based criterion that allow to use a smaller number of conformatons, thus reducing computation time and at the same time improving the prediction results.
The library introduces a number of new features and properties in the selection of conformes for side chain optimization
- The library is sorted by the efficiency of the conformer in fitting natural protein environments
- Because the library is sorted it can be resized to any desired number of conformers, which allows maximum flexibility in deciding conformational sampling
Library files:
- EBL, conformer library, with CHARMM22 atom and amino acid names: EBL_11-2011_CHARMM22.txt (37MB)
- EBL, conformer library, with PDB v2.3 atom and amino acid names: EBL_11-2011_PDB2.3.txt (37MB)
- EBRL, rotameric version of the library (standard bond distances and angles) with CHARMM22 atom and amino acid names: EBRL_11-2011_CHARMM22.txt (28MB)
- EBRL, rotameric version of the library (standard bond distances and angles) with PDB v2.3 atom and amino acid names: EBRL_11-2011_PDB2.3.txt (28MB)
- All four files, zip compressed: EBL.zip (26MB)
Tutorial for the creation of the library (first rough draft)
Requirements
a) a fine-grained conformer library in the MSL format to be ranked:
the EBL_11-2011_CHARMM22.txt file distributed with MSL may be used.
b) for CHARMM energetics, CHARMM topology and parameter ("top_all22_prot.inp" and "par_all22_prot.inp")
c) for hydrogen bonding, the hydrogen bond parameters file: the par_hbond_1.txt file distributed with MSL may be used.
d) for solvation, the solvation parameters file ("solvpar22.inp" or "solvpar.inp").
e) createEnergyTable program, distributed with MSL.
f) createEBL program, distributed with MSL
g) environments file and their pdb files (with CHARMM atom names), available here (see above link)
Note: Including the hydrogen bonding and solvation terms is optional with the createEnergyTable program.
Procedure
1) Use the createEnergyTable program to create a table of
energy values, with each row corresponding to an environment and each
column being a conformer from the fine-grained conformer library.
2) Use createEBL on the energytable produced in step 1 to sort the conformers.
3) Use the sorted library and run createEnergyTable again, now use
the sortedEnergyTable to determine the LEVELS for the sorted conformer
library.
createEnergyTable, programs options
| Required parameters |
|
| --pdbdir |
The directory containing the structures for all the cavities. Each structure file should be called <pdbId>.pdb |
| --envlistfile |
The file containing all the training environments |
| --rotlibfile |
The file containing the fine-grained conformer library |
| --charmmtopfile |
The CHARMM topology file. |
| --charmmparfile |
The CHARMM parameter file |
| --rawtablefile |
The output energytable file to be used with the createEBL program. For example:
4DOV A,104,ARG -2.30 -1.04 10.06 -7.22 8.36 ... |
| --sortedtablefile |
Similar to the energytable file but lists the
best energy obtained so far for each environment. For the environment
listed in the example the sortedtable file would look like the
following:
4DOV A,104,ARG -2.30 -2.30 -2.30 -7.22 -7.22 ...
This may be useful in determining the LEVELS for the sorted conformer library. |
| Optional Parameters |
|
| --hbondparfile |
The hydrogen bond parameters file. If not specified , no hydrogen bond terms will be used in the energy calculations. |
| --solvparfile |
The solvation parameters file. If not specified, no solvation terms will be used in the energy calculations. |
| --solvent |
The solvent to be used . Defaults to water if a solvation parameters file is specified. |
| --cuton |
The cutoff inner limit for non bonded energy
terms. Default is 9.0A. The limits are important to keep memory usage
under control and are sufficiently accurate for creating the energy
table. All non bonded interactions within <cuton> distance are
used unweighted. All interactions between <cuton> and
<cutoff> are weighted using a switching function. All
interactions between <cutoff> and <cutnb> are stored but add
zero energy to the calculations. All interactions greater that
<cutnb> are not stored (considered in energy calculations). Note
that all distances are as measured on the initial molecule, so as the
sidechains in the environments are remodeled, interactions may move
between the three states mentioned above. |
| --cutoff |
The cutoff outer limit for non bonded energy terms. Default is 10.0A. |
| --cutnb |
The cutoff limit for creating for non bonded energy terms. Default is 11.0A. |
| --cuthb |
The distance beyond which no hydrogen bonding interactions are considered. Defaults to 10.0. |
| --vdwrescalingfactor |
The vdw radii may be rescaled using this option. (expressed as a fraction, defaults to 1.0) |
| --configfile |
All the options to this program may be specified in a text file (omit the "--"). |
| --weight <term1,weight> --weight <term2,weight> |
The weights for each supported energy term may be specified. For example
--weight CHARMM_BOND,0.5 --weight CHARMM_VDW,0.75
By default all used energy terms have weight = 1.0 |
createEBL, programs options
| Required parameters |
|
| -resname |
The amino acid type of the cavities. |
| --energytablefile |
The "raw" energy table file generated by createEnergyTable program. |
| --rotlibfile |
The "unsorted" rotamer library used to create the energytable. |
| --outputrotlibfile |
The name of the file that will contain the result i.e) the sorted conformer library. |
| Optional Parameters |
|
| --configfile |
All the options to this program may be specified in a text file (omit the "--"). |
| --numrotstosort |
The number of conformers to be sorted. The first <numrotstosort> conformers are considered and the rest are ignored. |
| --offset |
An environment is satisfied by a conformer if
the energy for the conformer is less than (the best energy obtained by
all the conformers + offset). Default values for the offset are
ALA = 1.0;
ARG = 8.0;
ASN = 1.5;
ASP = 1.5;
CYS = 1.0;
GLN = 4.0;
GLU = 4.5;
HSD = 2.5;
HSE = 2.5;
HSP = 6.5;
ILE = 1.0;
LEU = 1.0;
LYS = 5.0;
MET = 3.0;
PHE = 1.5;
SER = 2.0;
THR = 1.0;
TRP = 3.5;
TYR = 2.0;
|
Material for the creation of the library
|
